Title: About the identification result.
Post by: liyong189108 on January 13, 2021, 09:28:02 AM
Post by: liyong189108 on January 13, 2021, 09:28:02 AM
Hi All,
I found something wierd about the identification result of my LC-MS/MS profiling data, The identification score cut off was set as 90% (see the attached screen shot) and a list of metabolites were identified. However, the annotation seems performed only based on MS1 ions. All identified metabolites in the exported alignment result table were annotated as false "MS/MS matched" while the total scores were mostly more than 90 (see attached table). I have checked some of the annoted candidates and make sure that the MS2 spectral was not matched well with the reference spectral. I can't understand the scoring rules because I think the candidate MS/MS spectral should matched very well with the library spectral when a high score was given. Could you please help me to remove this confusion?
Thanks
Yong Li
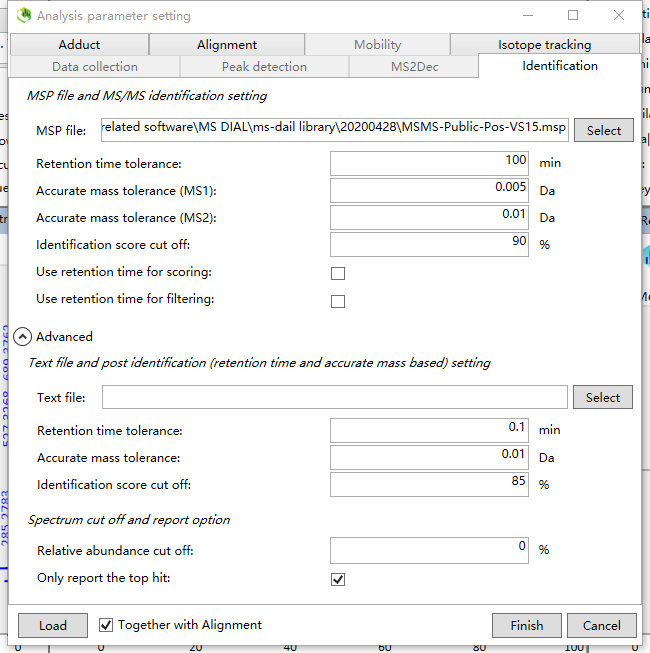
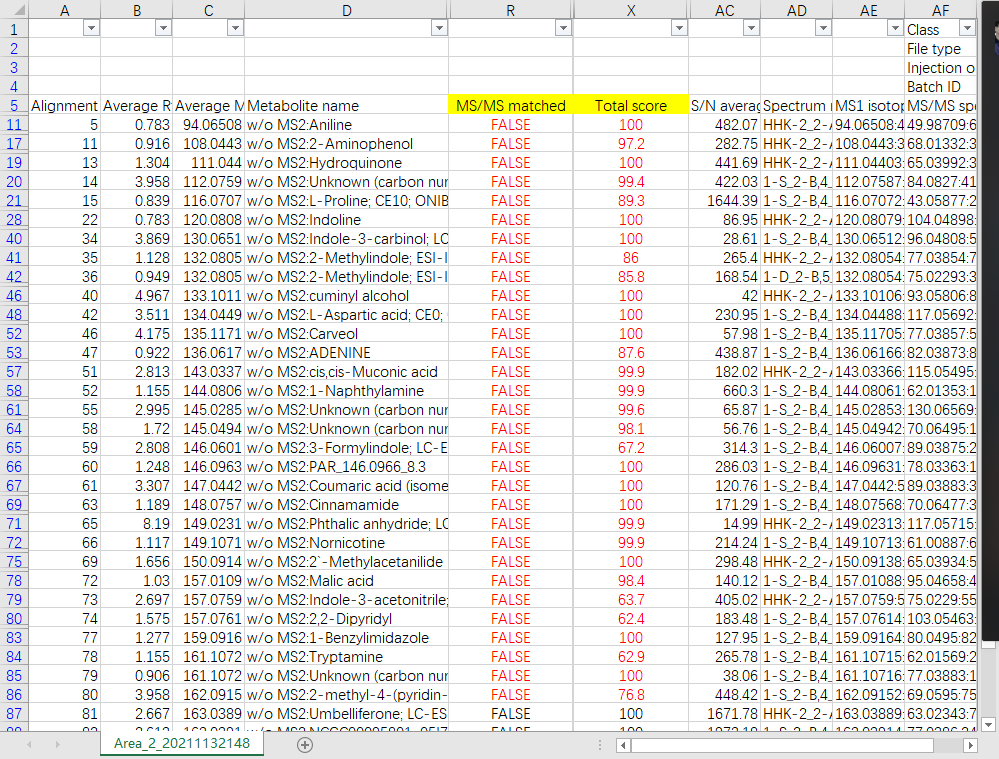
I found something wierd about the identification result of my LC-MS/MS profiling data, The identification score cut off was set as 90% (see the attached screen shot) and a list of metabolites were identified. However, the annotation seems performed only based on MS1 ions. All identified metabolites in the exported alignment result table were annotated as false "MS/MS matched" while the total scores were mostly more than 90 (see attached table). I have checked some of the annoted candidates and make sure that the MS2 spectral was not matched well with the reference spectral. I can't understand the scoring rules because I think the candidate MS/MS spectral should matched very well with the library spectral when a high score was given. Could you please help me to remove this confusion?
Thanks
Yong Li
Title: Re: About the identification result.
Post by: Hiroshi Tsugawa on February 07, 2021, 05:15:51 AM
Post by: Hiroshi Tsugawa on February 07, 2021, 05:15:51 AM
Hi,
sorry for this confusing result in the current msdial output.
In processing tandem mass spectral data, there are two ways for annotation scoring.
(1) based on RT, isotope, m/z accuracy, and MS/MS similarity
(2) based on RT, isotope, m/z accuracy.
Both scores are normalized (in metabolomics project) to 100 as the maximum score.
The problem is
(A) when a peak is annotated by the first criteria, the annotation will be performed by the second criterion.
(B) the second criterion was basically for the peak that does not have MS/MS spectrum information. However, it will be also applied to the peak having MS/MS spectrum if the peak is not annotated by the first criterion.
If the peak is annotated by the second criterion, the "w/o MS2: " tag is assigned.
The current problem is that the annotation scores of first and second criteria are normalized to 100 in both cases.
>>I think the candidate MS/MS spectral should matched very well
Check your m/z accuracy for ms1 and ms2. As a trial, please use a wider mass tolerance for ms1 and ms2.
Thanks,
Hiroshi
sorry for this confusing result in the current msdial output.
In processing tandem mass spectral data, there are two ways for annotation scoring.
(1) based on RT, isotope, m/z accuracy, and MS/MS similarity
(2) based on RT, isotope, m/z accuracy.
Both scores are normalized (in metabolomics project) to 100 as the maximum score.
The problem is
(A) when a peak is annotated by the first criteria, the annotation will be performed by the second criterion.
(B) the second criterion was basically for the peak that does not have MS/MS spectrum information. However, it will be also applied to the peak having MS/MS spectrum if the peak is not annotated by the first criterion.
If the peak is annotated by the second criterion, the "w/o MS2: " tag is assigned.
The current problem is that the annotation scores of first and second criteria are normalized to 100 in both cases.
>>I think the candidate MS/MS spectral should matched very well
Check your m/z accuracy for ms1 and ms2. As a trial, please use a wider mass tolerance for ms1 and ms2.
Thanks,
Hiroshi